
APC RESISTANCE UNIVERSITY
Introduction | Biochemistry | Clinical Aspects | Key Reports | PubMed Abstracts | References | Assay Methods | APC Resistance Phenotyping | Factor V Mutation Screening | FAQ
BIOCHEMISTRY
Initiation and regulation of blood coagulation
In order to prevent dangerous blood loss following vascular injury, the hemostatic system is called into action (Figure 3). Within seconds of injury the damaged vessel contracts and circulating, disc-shaped cell components called thrombocytes or platelets are activated and start to adhere to the site of injury. The activated platelets then aggregate to form a loose plug that reduces or temporarily stops the bleeding. The activated platelets also release a large number of molecules that accelerate platelet plug formation and begin the process of wound healing. Blood coagulation is triggered simultaneously with these events. This major biochemical process takes place at the surface of negatively charged phospholipid membranes provided by activated platelets and damaged cells. It results in the localized and timely formation of a fibrin matrix that stabilizes the platelet plug and seals the bleeding vessel. The fibrin clot is eventually lysed by the fibrinolytic system at the completion of the healing process.
The coagulation cascade

Although the cascade/waterfall model of blood coagulation has been modified during recent years it is still largely valid.17-18 The current scheme as seen in figure 4, involves a series of proteolytic reactions, in which inactive coagulation factors in a precursor or zymogen form are activated by one or more cleavages. Several of the activated coagulation proteases form complexes with their specific cofactors on the phospholipid surface, amplifying their activation of subsequent zymogens.
There are two activation pathways in the coagulation cascade, the intrinsic and the extrinsic pathway. The intrinsic pathway involves components intrinsic to whole blood, whereas the extrinsic pathway includes an extrinsic (subendothelial) activating component called tissue factor. Both pathways involve a number of plasma proteins as listed in Table 1. Most of the coagulation factors are zymogens of trypsin-like serine proteases that cleave arginyl peptide bonds with high specificity. Several proteins, including factors II (prothrombin), VII, IX and X, protein C and protein S, are subjected to vitamin K-dependent g-carboxylation of glutamic acid residues during their synthesis in the liver. This unique amino acid modification allows the proteins to bind calcium ions necessary for phospholipid binding and thereby to participate efficiently in multimolecular complexes in the coagulation cascade.17-18 Inhibition of g-carboxylation using vitamin-K-antagonistic drugs such as warfarin is a commonly used approach for anticoagulant treatment.
Initiation of blood coagulation
The extrinsic pathway is now accepted as the major activation route for blood coagulation in vivo.17 It becomes activated when disrupted tissue and activated monocytes exposes tissue factor to the bloodstream.19 Tissue factor forms a complex with factor VII, which becomes activated and then activates factors IX and X. The intrinsic pathway is initiated by the exposure of ‘contact’ factors in plasma (i.e. factor XII, HMW kininogen and prekallikrein) to a negatively charged surface, such as connective tissue in vivo or glass in a test tube.20 The two pathways converge on factor X to a common pathway, leading to the conversion of prothrombin into the key coagulation enzyme, thrombin.21
Table 1a. Blood coagulation factors
| Factor | Synonym | M.W. [kDa] | Plasma level [mg/ml] | Function |
| I | Fibrinogen | 340 | 3000 | Structural |
| II | Prothrombin* | 72 | 100 | Protease zymogen |
| III | Tissue factor | 37 | – | Cofactor/initiator |
| IV | Calcium | – | – | – |
| V | Proaccelerin | 330 | 10 | Cofactor precursor |
| VI | – | – | – | – |
| VII | Proconvertin* | 55 | 0.5 | Protease zymogen |
| VIII | Antihemophilic factor | 330 | 0.1 | Cofactor precursor |
| IX | Christmas factor* | 55 | 5 | Protease zymogen |
| X | Stuart-Prower factor* | 55 | 10 | Protease zymogen |
| XI | Thromboplastin antecedent | 160 | 5 | Protease zymogen |
| XII | Hageman factor | 80 | 30 | Protease zymogen |
| XIII | Fibrin-stabilizing factor | 320 | 30 | Protransglutaminase |
| – | Prekallikrein | 85 | 40 | Protease zymogen |
| – | HMW kininogen | 120 | 80 | Cofactor/activation |
| – | Von Willebrand factor | >1500 | 10 | Adhesion, carrier protein |
| 220 kDa subunits |
Table 1b. Regulatory proteins of blood coagulation and fibrinolysis
| Name | M.W. [kDa] | Plasma level [mg/ml] | Function |
| Tissue factor pathway inhibitor | 40 | 0.1 | Protease inhibitor |
| Antithrombin | 58 | 150 | Protease inhibitor |
| Heparin cofactor II | 66 | 90 | Protease inhibitor |
| Protein C* | 62 | 4 | Protease zymogen |
| Protein S* | 78 | 20 | Cofactor |
| Thrombomodulin | 60 | – | Receptor |
| Protein C inhibitor | 57 | 5 | Protease inhibitor |
| Plasminogen | 92 | 200 | Protease zymogen |
| t-PA | 70 | 0.005 | Protease |
| u-PA | 54 | 0.008 | Protease |
| PAI-1 | 52 | 0.02 | Protease inhibitor |
| PAI-2 | 47 | <0.005 | Protease inhibitor |
| Plasmin Inhibitor | 70 | 70 | Protease inhibitor |
* Vitamin K-dependent protein containing g-carboxyglutamic acid.Data was compiled from: Nomenclature of quantities and units in Thrombosis and Haemostasis 71, 375-394 (1994). and Mosher et al, Clin Cardiol 13, VI-5-11 (1990).
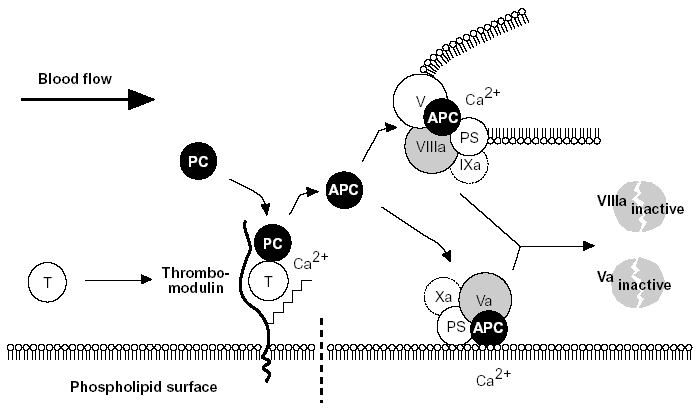
Figure 5. The protein C anticoagulant pathway.
Thrombin escaping from a site of vascular injury binds to its receptor thrombomodulin (TM) on the intact cell surface. As a result,thrombin loses its procoagulant properties and instead becomes a potent activator of protein C. Activated protein C (APC) functions as a circulating anticoagulant, which specifically degrades and inactivates the phospholipid-bound factors Va and VIIIa. This effectively down-regulates the coagulation cascade and limits clot formation to sites of vascular injury. The activity of APC is potentiated by two cofactors, protein S and native (non-activated) factor V. Protein S functions as a cofactor in the degradation of factor Va and VIIIa. Native factor V acts in synergy with protein S as a cofactor in the degradation of factor VIIIa. Thus, factor V has dual roles: one as anticoagulant in its native form and the other as an procoagulant after its activation. APC is slowly neutralized by circulating inhibitors. Thrombin bound to TM is eventually inhibited by antithrombin or removed through endocytosis of the thrombin/TM complex.
Symbols: T= thrombin, PC= protein C, PS= protein S.28
The serine protease thrombin converts circulating fibrinogen into clot-forming fibrin molecules and activates the transglutaminase, factor XIII, which stabilizes the fibrin matrix through covalent cross-linking. Thrombin also stimulates cellular hemostasis and coagulation through positive feedback, by activating platelets and the two circulating non-enzymatic cofactor proteins, factor V and factor VIII.22-23 All these feedback activations by thrombin lead to an explosive amplification of the coagulation cascade and rapid clot formation.
Thrombin regulation
It is evident that the autocatalytic nature of thrombin could clot the blood content of a person within minutes if uncontrolled. In humans, the necessary control involves two aspects, i.e. inhibition of thrombin already formed and prevention of further thrombin generation. Direct thrombin inhibition is provided primarily by circulating serine protease inhibitor, antithrombin,25 whereas the crucial prevention of thrombin generation is provided indirectly by thrombin itself. This self-regulating function of thrombin is expressed in its binding to thrombomodulin, a specific, high-affinity receptor protein located on undamaged (intact) endothelium.26,27
The protein C anticoagulant pathway
On binding to thrombomodulin, thrombin loses all its procoagulant properties. Instead, it becomes a potent activator of protein C, the ‘prima ballerina’ of the protein C embolic disorder observed in infants with severe protein C or protein S deficiency.
Natural substrates of APC: factors Va and VIIIa
Factors V and VIII are two large, relatively unstable, plasma proteins of about 330 kDa, with similar structure and function.22-24 Factor V is an essential component for the rapid conversion of prothrombin to thrombin, whereas factor VIII is needed to accelerate the activation of factor X to factor Xa. The essential role of these non-enzymatic cofactor proteins in hemostasis is evidenced by the severe bleeding tendency associated with their deficiency.22,33 Both factors are synthesized mainly in the liver and circulate in plasma as inactive molecules with little or no procoagulant activity. A unique feature of factor VIII is that it circulates in a stabilizing, non-covalent complex with the von Willebrand factor, an adhesive protein that is important for the proper function of platelets.23 The plasma concentration of factor V is about 10 mg/ml, which is up to a 100-fold higher than that of factor VIII (0.1-0.2 mg/ml).28 About 20% of the total amount of factor V in blood is synthesized by megacaryocytes and stored in platelets. This stored form of factor V is released in conjunction with platelet activation and has an important role in normal hemostasis. The genes for factor V and VIII are located on chromosomes 1 and X respectively, coding for mature, singlechain proteins of roughly 2200 amino acids. Prior to secretion into the bloodstream, the factor VIII molecule is processed to a calcium ion-linked heterodimer, whereas factor V circulates as a single-chain protein.22-23 Computer- aided comparison of the primary amino acid sequence of factors V and VIII reveals a high degree of homology, with an overall identity of about 30%.24 Both proteins contain several types of similar internal repeats, termed A1-A2-B-A3-C1-C2.
Proteolytic activation of factor V and factor VIII
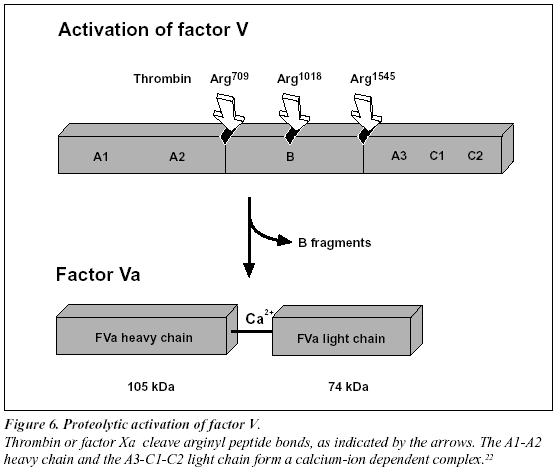
Factors V and VIII are activated through limited proteolysis by thrombin or factor Xa.22-23 During its activation, factor VIII is released from the protective influence of the von Willebrand factor and converted to a calcium ionanticoagulant pathway (Figure 5). The activated protein C (abbreviated APC) is a serine protease that rapidly downregulates thrombin generation, by cleaving and inactivating the phospholipid-bound activated forms of coagulation factors V and VIII (factor Va and factor VIIIa). APC in turn is only slowly neutralized by three inhibitors, protein C inhibitor, trypsin inhibitor and a2-Macroglobulin. The relatively long half-life of APC in vivo (15-20 minutes) is a prerequisite for its function as a circulating anticoagulant.
Protein S
The anticoagulant activity of APC is potentiated and supported by protein S, a non-enzymatic plasma protein.27-28 Its mechanism of action is not yet completely understood, but protein S has been shown to promote the binding of APC to phospholipid surfaces, and to remove the factors Xa and IXa-mediated APC protection of factors Va and VIIIa respectively.28 Protein S has also been reported to stimulate the inactivation of factor Va 20-fold by specific acceleration of the cleavage at Arg306, one of the three cleavage sites for factor Va inactivation (See Figure 7).287 Finally, it may have an APC-independent anticoagulant activity, by inhibiting prothrombin activation through direct interaction with factor Va and factor Xa.28 About 60% of the protein S in plasma is bound to C4bBP, a regulatory protein of the classic complement system and is not active as APC cofactor.
Anticoagulant role of factor V
Recently, it has been found that non-activated factor V in synergy with protein S functions as an APC cofactor in the degradation of factor VIIIa.29 This has been confirmed in other studies,30-31 although the view of intact factor V as an anticoagulant APC cofactor has recently been challenged. Instead, it has been suggested that the central Bdomain released on activation of factor V expresses the APC cofactor activity (Figure 5).32 The in vivo relevance of these findings awaits further investigation. Taken as a whole the protein C pathway constitutes an ingenious mechanism by which procoagulant thrombin attains anticoagulant properties in the absence of vascular injury.28 The physiological importance of this mechanism is demonstrated clinically by the massive thrombo-dependent trimer (A1, A2 and A3-C1-C2). The active factor V molecule is a dimer that consists of a heavy chain (A1-A2), non-covalently linked via calcium ions to a light chain (A3-C1-C2) (Figure 6). The activated factors V and VIII bind to negatively charged phospholipid in the presence of calcium and serve as cofactors/receptors for factors IXa and Xa respectively. The importance of these multimolecular complex assemblies, better known as the tenase and prothrombinase complexes, is evidenced by the over 100,000-fold increase in the combined rate of activation of factor X and prothrombin when compared to the activation catalyzed by their respective enzyme alone.24
APC inactivation of factors Va and VIIIa
The native form of circulating factors V and VIII are poor substrates for APC. In contrast, APC effectively cleaves and inactivates the phospholipid-bound activated form of these proteins. The inactivation of factor Va takes place through the APC-mediated cleavage in the heavy chain of the molecule of three peptide bonds at Arg506, Arg306 and Arg679 (Figure 7).34,285 Cleavage at Arg506 is needed for the efficient exposure of the cleavage sites at Arg306 and Arg679. The lipid-dependent cleavage at Arg306 appears to be the major inactivating cleavage site and results in a loss of about 80% cofactor activity, whereas cleavage at Arg679 is lipid-independent and is responsible for the loss of the remaining cofactor activity.34 Potential structural differences between platelet factor Va and plasma factor Va may influence the extent to which the cofactor is cleaved initially at Arg306.35 APC inactivates factor VIIIa by cleavages at Arg336, Arg562 and Arg734. The main loss of factor VIIIa cofactor activity is associated with the cleavage at Arg562.36
Molecular explanation of APC resistance
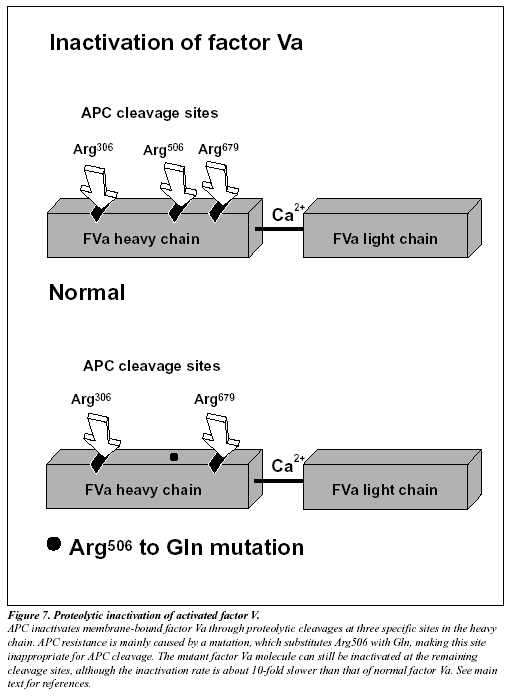
The initial observation that normal factor V mixed with APC resistant plasma was able to correct the APC response in a dose-dependent manner, suggested to several independent research groups that APC resistance was due to a defect in the factor V molecule.13,37-38 However, the precise molecular explanation was discovered first by a Dutch group led by R. Bertina.13 The APC resistance phenotype in this seminal study was linked to a single-point mutation in the factor V gene, which substitutes G (codon CGA) with A (codon CAA) at nucleotide 1691 in exon 10.13 This mutation replaces Arg (R) with Gln (Q) at position 506 in the factor V molecule, thus modifying one of the three APC cleavage sites (Figure 7). The mutant factor V molecule (abbreviated FV:Q506) expresses normal procoagulant activity when activated by thrombin or factor Xa, although its rate of inactivation is about 10-fold slower than that of normal factor Va.39-42 This “resistance” to degradation by APC allows for a longer duration of thrombin generation, which may be reflected by increased levels of coagulation activation markers such as prothrombin fragment 1+2, thrombin-antithrombin (TAT) complex and D-dimer.43-46 Recent data also suggest that a reduced ability to slow down thrombin generation may stabilize a blood clot by weakening the profibrinolytic effect of APC.48 An antifibrinolytic mechanism could thus be an additional factor contributing to the prothrombotic tendency observed in APC resistant patients. The fact that mutant FVa:Q506 can still be inactivated by APC cleavage at Arg306 and Arg679, might account for the relatively mild hypercoagulable state observed in APC-resistant individuals and help explain why additional genetic and/or acquired risk factors are required for thrombosis to develop.41
Heterogeneous phenotype
Most cases of inherited APC resistance are caused by the factor V mutation. However, as seen under “normals” in Figure 9 (page 18), the APC resistance phenotype is clearly heterogeneous, being influenced by other factors as well. Several reports have shown that about 10% (range 4-20%) of APC resistant cases among Caucasians, do not involve the FV:Q506 mutation.13,16,41,47 The cause of this type of APC resistance is not known but may be the result of acquired APC resistance or due to other genetic defects. Analogous to the FV:Q506 mutation, APC resistance could be explained by mutations at the APC cleavage sites of factor VIII, for example a mutation at Arg336 or Arg562. Although, to date no such mutations have been found.49
Summary
Protein C is a vitamin-K-dependent plasma proenzyme of a serine protease that plays a key role in the down-regulation of blood coagulation. It is activated in vivo by the thrombin-thrombomodulin complex on the surface of intact endothelial cells. Activated protein C (APC) functions as a circulating anticoagulant through proteolytic cleavage and inactivation of two critical, phospholipid-bound coagulation proteins, factors Va and VIIIa. The cleavages occur at three sites in the heavy chain of each proteins. The anticoagulant activity of APC is potentiated by protein S and non-activated factor V. APC resistance is mainly due to a point mutation (G to A at nucleotide 1691) in the factor V gene that predicts replacement of arginine at position 506 by glutamine in the factor V molecule. The mutation destroys one of the three APC cleavage sites, rendering the activated and procoagulant factor Va:Q506 partially resistant to APC-mediated degradation. About 90% of APC resistant cases can be explained by the factor V mutation.
ORDER: 1-800-524-52224 | SUPPORT: 1-800-447-3846 | ABOUT US | CONTACT US | HOME